Оглавление Введение Часть 1 Часть 2 Часть 3 Заключение
Глава 1 Глава 2 Глава 3 Глава 4 Глава 5 Глава 6 Глава 7 Литература
Глава 2
Инструментальное обеспечение гомеостатического устройства: химические превращения, норма реакции, катализ.
Все известные химические взаимодействия и превращения происходят при смешении или физическом контакте (столкновениях) реагентов самопроизвольно, при нагревании, участии катализаторов (катализ), действии света (фотохимические реакции), электрического тока (электрохимические, электродные процессы), ионизирующих излучений (радиационно-химические реакции), механических воздействиях (механохимические реакции), в низкотемпературной плазме (плазмохимические реакции) и т.д.
Единственным обязательным условием для их исполнения является факт наличия энергии, достаточной для преодоления так называемого «потенциального барьера», «разделяющего» исходное и конечное состояния системы, т.е. энергии активации[1] (рисунок 13).
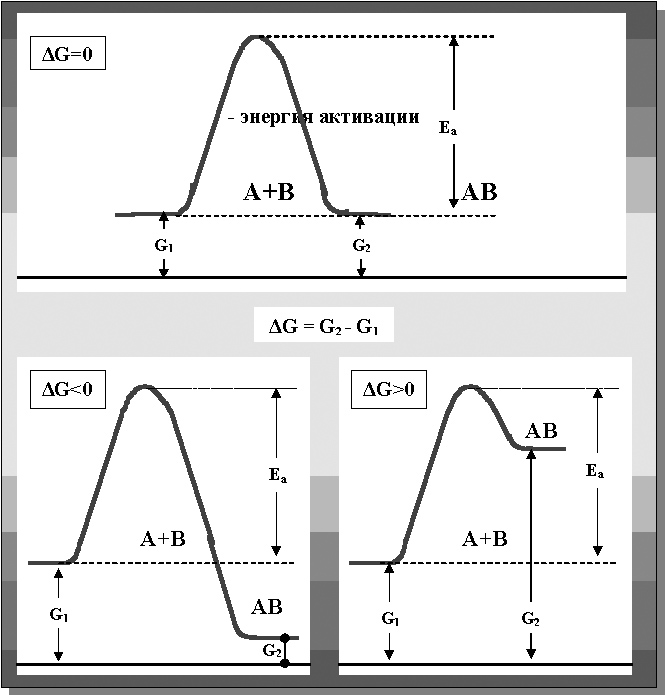
Рисунок 13. Свободная энергия и энергия активации при равновесных (ΔG=0),
экзо- (ΔG<0) и эндотермических (ΔG>0) реакциях
Самопроизвольные (первичные) химические реакции в отличие от ядерных реакций не связаны с изменением ни общего числа атомов в реагирующей системе, ни с изотопным составом химических элементов. Всё их действие происходит в полном соответствии закону сохранения действующих масс (например: А+В=АВ). И, если его (закон сохранения) выразить через константу равновесия[2] (Кр)[3]:
Кр=[A][B]/[AB]
где: [A] и [B] – концентрации реагентов
[AB] – концентрация продукта реакции,
то можно легко рассчитать изменение стандартной свободной энергии Гиббса[4] (DG°):
DG° = -RT ln Кр
где: R – газовая постоянная
T – абсолютная температура
характеризующей поведение химических реакций при нагревании и в полном соответствии с правилами Шелфорда и Либиха, представляющими в комплексе суть основы нормы исполнения химических реакций.
Фотохимические реакции происходят под действием света, исполняющего роль некоего энергетического донора, компенсирующего дефицит энергии, необходимый для преодоления энергетического барьера (энергии активации). Большинство из них это многостадийные процессы, начинающиеся с поглощения фотона молекулой, что приводит к образованию возбуждённого состояния[5]. Это требует сообщения энергии, приблизительно равной 1-9,5 эВ.
Состояние возбуждения весьма не продолжительно. В нём молекулы могут пребывать приблизительно 100 наносекунд (нс), но не более 10 пикосекунд (пс):
~10-7-10-8с, но не более 10-11с.
За это время они либо вступают в первичные химические реакции, либо возвращаются в исходное состояние, испуская квант света (флуоресценция, фосфоресценция).
Возбуждённые молекулы могут также образовываться при «безизлучательном» переносе энергии и при последовательном поглощении двух или нескольких фотонов[6], приводящих к образованию высоковозбуждённых молекул (t-молекулы) с энергиями
6,5-9,5 эВ,
тогда как одно-квантовые реакции (p-молекулы) происходят с энергиями, не превышающими 5 эВ[7]. Время жизни t-молекул несколько больше и составляет
~10-6-10-5с
у p-молекул:
~10-7-10-8 £ 10-11с.
К фотохимическим реакциям относят:
1. реакции Нориша - расщепление кетонов под действием света,
2. фотовосстановление, связанное с межмолекулярным и внутримолекулярным переносом электронов,
3. фотоизомеризацию - внутримолекулярные ионные или радикальные реакции, а равно синхронное перераспределение связей в молекуле[8],
4. фотополимеризацию цепного (полимеризационного) или ступенчатого (поликонденсационного) типа,
5. фотоокисление, как процесс межмолекулярного переноса электронов,
6. фотоперенос электронов от донора (восстановителя) к акцептору (окислителю),
7. фотопревращение радикалов: диссоциация, замещение, изомеризация, ионизация,
8. фотоаутомерию прототропного типа, связанную с переносом протонов, и анионотропного типа, т.е. перемещение анионов, а также кольчато-цепного типа действия, завершающегося замыканием и размыканием циклов,
9. эксиплексы, это возбуждённые молекулярные комплексы, связанные по донорно-акцепторному типу взаимодействия, и перераспределяющие или переносящие свой заряд.
В плане нашего изложения, фотохимические процессы чрезвычайно важны для понимания эволюции систем управления гомеостатическими состояниями прямого типа действия. Ведь именно они представляют собой суть образец механизма акцепта реальных физических (волновых) полей и потоков релятивистского свойства, поступающих от высших иерархов гомеостаза в нерелятивистский мир следующего уровня организации: от высокоорганизованного с видимым преобладанием массовых свойств до элементарного с преобладанием волновых качеств.
Таким образом, нам предоставляется уникальная возможность утверждения центральной роли волновых процессов в организации жизни всех участников гомеостатического мира, построенных на принципах дуализма и сохранения баланса структурно-функциональных и квантовомеханических свойств в реальном мире, включая:
1. способность акцепта энергии и информации через возбуждение;
2. способность преобразования сверхвысоких частот в более низкие путём их аккумуляции (на примере двухквантовых процессов) и трансформации (на примере фотосинтетических процессов);
3. способность аккумуляции энергии высоких частот в энергию молекулярных и межмолекулярных связей (на примере фотосинтеза).
Электрохимические реакции (электродные процессы) рассматриваются современной физикой, как правило, только с позиций некого прикладного свойства, характеризуя их способность переноса зарядов через границу сред (между электродом и электролитом), всегда идущим в двух направлениях: катодном и анодном. При этом катодные процессы связывают с переносом электронов из металла на молекулы или ионы реагирующих на электроде веществ, а анодные - с окислением реагирующих веществ, сопровождающемся переходом электронов на металл, либо катионов металла в раствор.
Безусловно, эти обстоятельства имеют место и абсолютно верны в своём изложении, но носят только частный характер, освещая лишь некую исторически обособленную сферу как бы «искусственных» человеческих интересов: «электрику», электродинамику, электростатику, электрохимию, электронику.
А ведь по своему существу, электрохимические процессы присутствуют везде, во многом являясь определяющими. От элементарного, несущего электрический заряд, до сверхмощных грозовых зарядов, наполняющих атмосферу планетарной биосферы. От электростатических отношений до электромагнитных потоков. От стехиометрии электростатического сродства субстратных центров до специфики ионизированных потоков.
Именно они формируют так называемые диффузионные и трансмембраннные потенциалы, обеспечивающие сопряжение разнообразных диффузных процессов с фосфорилированием (протонное фосфорилирование, ионное фосфорилирование), составляющих суть энергообразующих (энергоснабжающих) биохимических реакций.
Именно они обеспечивают конформационную подвижность (лабильность) третичной, четвертичной и пятеричной молекулярных структур, активностей субстратных центров и рецепторных зон биомолекул, обслуживающих транспортные, каталитические, иммунные и иные стехиометрические процессы, составляющие суть свойств и проявлений электрофизиологических взаимоотношений высокоорганизованной материи, сохраняя при этом дееспособным механизм сохранения чувствительности участников нерелятивистских процессов к релятивистским скоростям когерентного им волнового (или электрического) мира.
Основным уравнением электрофизиологии, формализующим отношения материального, термодинамического и электрического мира, о котором мы говорим, является, безусловно, уравнение выдающегося немецкого физика, профессора Берлинского университета и директора Физического института Вальтера Германа Нернста[9] (1864-1941):
Dy = (RT/F)×ln (c1/c2),
где: Dy - разность потенциалов
R - газовая постоянная
T - абсолютная температура
F - число Фарадея
c1 и c2 - ионные концентрации с двух сторон разделительной мембраны
Основным же уравнением, регламентирующим электрохимические силы, является электрохимический потенциал, аналог химического потенциала (mia) для систем, содержащих заряженные частицы или оперирующих ими:
mia = mia + Zi×F×ja,
где: mia - химический потенциал частицы в фазе
Zi - заряд частицы с учётом её знака
F - число постоянная Фарадея
ja - электрический потенциал
mia = (¶G/¶ni)T, p, nj¹i
где: G – значение энергии Гиббса
i – заряженный компонент
a - фаза, содержащая заряженный компонент i
- число молей компонента i
Электрохимический потенциал можно определить также как умноженную на постоянную Авогадро работу (A) переноса (m) заряженной частицы (i) из бесконечно удалённой точки с нулевым потенциалом внутрь фазы a:
mia = 6,022045(31)×1023 моль-1 ×m×A
Радиационно-химические реакции происходят вследствие поглощения веществом жёстких ионизирующих излучений с длиной волны ~8×10-9-10-10с или 8,0-0,1 нм. Потоки частиц и электромагнитных квантов, взаимодействуя со средой, приводят к ионизации её атомов и молекул (эффект ионизирующего излучения). Ионизирующим является рентгеновское и g-излучение, потоки a-частиц, электронов, позитронов, протонов и нейтронов.
Заряженные частицы ионизируют среду непосредственно при столкновении с её атомами и молекулами (первичная ионизация). Выбиваемые при этом электроны, если они обладают достаточно большой энергией (эффект линейной передачи энергии), также могут ионизовать (вторичная ионизация):
1. в случае быстрых нейтронов ионизация обусловлена ядрами отдачи или другими частицами, возникающими при взаимодействии нейтронов со средой;
2. в случае ионизации фотонами рентгеновского или g–излучений ионизация может быть непосредственной – первичной (фотоионизация), а также, в большей степени, вторичной, обусловленной электронами, образовавшимися при воздействии фотонов на вещество.
Как и при фотохимических реакциях, первичные продукты – ионы и возбуждённые короткоживущие молекулы (время жизни этих продуктов ~10-8с), реагируя с молекулами среды или друг с другом, приводят к образованию относительно долго живущих свободных радикалов (R·), ион-радикалов (R+, R-) и других относительно стабильных продуктов:
1. в газовой среде, например, из кислорода образуется озон; из предельных углеводородов – водород и сложная смесь углеводородов различного строения;
2. в облучённой воде и разбавленных растворах образуются гидратированные электроны, радикалы ОН- и Н+; водород, Н2О2, а также ионы Н3О+
Механохимические реакции инициируются или ускоряются каким-либо механическим действием. Например, в твёрдых веществах при измельчении, действии ударных волн или высокого давления (»10Гпа = 103 Н×мм2 = 0,99×104 атм. = 1,01×104 кг-с×см2) в сочетании с деформацией сдвига (эффект Бридж мена). В жидкостях при кавитации (образование пузырьков, например, под действием ультразвука, растягивающих напряжений), действии сдвиговых напряжений на растворы или расплавы полимеров.
В водных средах при адиабатическом захлопывании кавитационных пузырьков пары воды диссоциируют на радикалы Н+ и ОН-, которые инициируют окислительно-восстановительные реакции, полимеризацию и т.д.
В твердых телах, полимерах и кристаллах механохимические реакции могут быть вызваны развитием деформаций в напряжённом материале, их разрушением и трением. Разрушение и трение, в свою очередь, вызывают кратковременное возбуждение атомов и молекул в приповерхностном слое вещества с образованием свободных радикалов R·.
Плазмохимические реакции представляют собой термохимические процессы взаимодействия ионизированных веществ равных по плотности положительных и отрицательных зарядов, осуществляемые в низкотемпературных условиях, когда средняя энергия ионов находится в пределах ~0,1-50 эВ. При этом и реагенты и продукты могут находиться в возбуждённом состоянии.
Важно, что в любом из обозначенных вариантов, баланс возбуждённых и невозбуждённых молекул будет всегда регулироваться законом сохранения энергии:
DН* = DН – (Е – Е¢)
где: DН* - энтальпия реакции в возбуждённом состоянии
DН - энтальпия реакции в основном (не возбуждённом) состоянии
Е - энергия возбуждения исходного вещества
Е¢ - энергия возбуждения первичного продукта
При этом инструментом исполнения, а соответственно, объектом управления является скорость достижения означенного баланса, т.е. скорость реакции.
Скорость реакции
Абсолютно все химические процессы протекают хотя и типически, но собственными индивидуальными путями. Каждый из них включает в себя почти мгновенное превращение одной или нескольких молекул субстрата в одну или несколько молекул продукта. Чтобы разобраться в ходе таких реакций, нужно уметь понять путь, по которому пойдут основные превращения, и скорость, с которой они осуществятся при любом наборе условий нормы реакции. В частности, рассматривая привычную уже нам реакцию ассоциации, при которой две молекулы реагентов, взаимодействуя друг с другом (бимолекулярная реакция[10]), образуют некое качественно новое объединение (вещество):
А+В=АВ
В этом случае, скорость реакции будет пропорциональна скорости, с которой сталкиваются молекулы А и В с частотой столкновений n, которую можно представить себе как число таких столкновений (соударений) А и В, происходящих в единице объёма и за единицу времени.
Если концентрацию [В] и продуктов реакции [AB] условно поддерживать постоянными, то n (частота столкновений) будет пропорциональна локальной концентрации [А] реагента А, поскольку каждая молекула А будет соударяется с молекулами В вещества с соответствующей концентрации [А] реагента А скоростью V. Логика рассуждений показывает, что и частота n эффективных соударений А и В должна быть также пропорциональна концентрации [В] реагента В и концентрации [AB] продукта AB, т.е.:
n = n0 [А][В][AB]-1,
где n0 – частота столкновений при концентрациях [А] и [В] равных единице. Тогда общая скорость реакции может быть выражена уравнением:
Скорость = Рn = Рn0[А][В][AB]-1,
где: Р – эффективная доля соударений А и В, приводящая к реакции.
Если доля молекул реагентов с энергией, большей, чем энергия активации Е, выражающейся как:
еxp(-E/kT),
где: Е – энергия на пару реагирующих молекул,
Т – абсолютная температура,
k – константа Больцмана
тогда:
P = s еxp(-NE/NkT) = s еxp(-E/RT),
где: Е – энергия активации в Дж×моль-1
N – число Авогадро
R – газовая постоянная (R = Nk).
В результате, общая скорость реакции может быть выражена как
Скорость V = s еxp(-E/RT)[А][В][AB]-1
Если при этом температура и давление будут поддерживаться постоянными, то уравнение может быть записано следующим образом:
Скорость V = k [А][В][AB]-1
где: k - константа: k = n0 s еxp(-E/RT)
Иными словами, скорость химической реакции, являясь показателем изменения концентрации реагирующих веществ во времени, выступает как генеральный инструмент эффективного исполнения управляющих условий норм реакции и может быть рассчитана как по убыли концентраций исходных реагентов
(-d[A]/dt) или (-d[В]/dt),
так и по возрастанию концентрации продуктов реакции:
(d[AB]/dt)
за установленный период времени. При этом скорость реакции всегда будет прямо пропорциональна произведению концентраций реагентов, индивидуальной константе скорости реакции[11] и обратно пропорциональна концентрации продуктов реакции:
Скорость = k1[A][B][AB]-1
Вместе с тем, число молекул, участвующих в реакции, даёт возможность определить её порядок (порядок реакции[12]):
1. Реакции нулевого порядка – это такие реакции, скорость которых не зависит от концентрации реагентов;
2. Реакции первого порядка, протекающие со скоростью, прямо пропорциональной концентрации реагирующих веществ и конечного продукта реакции в каждый данный момент времени и
3. Реакции высоких порядков, имеющих скорости, пропорциональные высоким степеням концентраций реагентов и продуктов реакции:
Скорость V = k1[A]а[B]b[AB]-1
Норма реакции
Из вышеизложенного следует, что изначальное существо нормы реакции есть набор неких оптимальных условий, концентраций реагентов, катализаторов, каталитических и других управляющих систем для сбалансированного исполнения термодинамически выгодных химических превращений в полном соответствии законам сохранения массы, вещества и энергии.
Исполнение требований нормы реакций и когерентность управляющей информации прямого действия осуществляется посредством эффекта смещения условий, концентраций реагентов, активностей участников или получаемых продуктов искомой «нормы реакции», равно связанный с возбуждением молекулярных орбиталей, индукцией, резонансами или интерференцией на элементарном, молекулярном или ином де Бройля уровне, сопровождающийся изменением пространственной конфигурации, структуры, активностей участников, скорости исполнения реакции.
Смещение же условий для исполнения требований нормы реакций в общем балансе отношений их участников можно назвать базовым управляющим фактором, требующим изменений скорости таких превращений, – «скорости реакций».
В связи с этим, говоря о механизмах управления химическими превращениями нельзя не учитывать, что они подчинены не только элементарным равновесиям, составляющим суть субстратного регулирования, но и принципам ионного регулирования эффективностей и скоростей исполнения условий «норм реакций».
Это более медленный процесс, чем процесс, связанный с эффектами прямого внешнего и внутреннего действия, но его статистическая природа обеспечивает стабилизацию условий исполнения «норм реакций» при взаимодействиях любых участников, обладающих реальными физическими зарядами, статическими, электростатическими, магнитными и электромагнитными полями, облегчающими процессы обмена веществом, энергией и информацией.
Следует также выделить подчиненность поведения «норм реакций» факторам прямого физического действия: магнитных, электромагнитных и гравитационных условий среды, а также влияния температуры и давления, составляющих единый принцип физического регулирования тех же эффективностей и скоростей экзо- и эндотермических «норм реакций».
Поднимаясь вверх по эволюционной или, иначе говоря, по гомеостатической лестнице, такая подчиненность сохранена повсеместно и может быть лишь усилена (а также разнообразиться) свойствами компартментации (локализации) и стехиометрии (пространственной ориентации). Причем такое усиление, если присмотреться к нему внимательнее, всё больше и больше усиливает регуляторные возможности управляющих систем субстратного, ионного и физического регулирования, сохраняя неизменными принципы обратной связи и принцип термодинамической рентабельности.
При этом, наиболее эффективными, а в ряде случаев наиболее оперативными элементами управления становятся внешние факторы воздействия (внешняя среда), возможность манипулировать субъектами «реакций» индуктивно, быстро и эффектно используя комбинации прямой (непосредственное действие), обратной (опосредованное действие) положительной (усиление) и отрицательной (ингибирование) связей[13].
Субстратное регулирование скорости реакции
На основании вышеизложенного, а также используя принципы «Теории возмущений», постараемся идентифицировать последовательный ряд ожидаемых событий и их участников при исполнении нормы реакции А+В=АВ, где управляющим её инструментом является скорость реакции, а фактором управления (активации или ингибирования) концентрации участников и продукты реакции:
1. инструмент исполнения требований нормы реакции А+В=АВ - скорость реакции (V)
V = k1[A]а[B]b[AB]-1;
2. факторы возмущения: реагенты [A] и [B] в виде вещества, энергии (E), информации[14] (I);
k1 = [A]а[B]b[AB]-1
3. фактор ингибирования: [AB] в виде вещества;
4. акцепт вещества [A][B], энергии (E) и информации (I);
5. возмущение молекулярных орбиталей[15] участников реакции A и B:
6. модуляция информации, выраженная во взаимодействии реагентов A и B, появление продукта [АВ];
7. изменение свойств, качества или реактивности участников акцепта A+B и продукта [АВ];
8. изменение баланса «нормы реакции»
[A][B][A,B,E или I]/[АВ] > constant k
9. изменение скорости реакции
V([A][B])/[ АВ] = k < V([A][B][A,B,E,I])/[АВ] > k
10. восстановление баланса согласно требованиям нормы реакции
[A][B])/[АВ]= constant k
11. восстановление скорости исполнения нормы реакции
V([A][B])/[АВ] = k
Именно эти события и названные в них участники определяют суть механизмов субстратного регулирования скорости реакции, как базового инструмента в исполнении норм любой химической реакции.
Ионное регулирование скорости реакции
Если механизмы субстратного регулирования, связанные с доминирующим влиянием концентраций участников (реагентов и продуктов) химических реакций и превращений управляют скоростью исполнения норм реакций, то процессы ионного регулирования балансируют её оптимум, используя для этого аналогичное уравнение, соотносящее концентрации ионных отношений диссоциирующего продукта:
АВ « kаb [А+] [В-],
pA+ = pkab + lg [В-][АB]-1
pВ- = pkаb + lg [A+][АB]-1
где: kаb – константа диссоциации
pA+ - парциальное давление искомого катиона
pВ- - парциальное давление искомого аниона
Но, если в механизм субстратного регулирования заложен принцип непосредственного регулирования скорости реакции, то механизм ионного регулирования направлен на ингибирование или активацию скорости реакции через изменение (уменьшение или увеличение соответственно) энергии активации самой реакции. Иными словами, ионные отношения исполняют при этом роль некоего катализатора (эффектора) основного химического процесса, подчиняясь, между тем, собственному закону равновесия:
kа = [AB] [A+]-1[B-]-1
В большей степени это может быть отнесено и к иным ионам, выступающим в качестве эффекторов исполнения скоростей целого ряда химических и, в особенности, биохимических реакций. В частности, это касается концентрации водородных ионов [Н+] в водных растворах (pН+).
Молекулы воды диссоциируют Н2О « 2(Н+ + ОН-) в соответствии с константой диссоциации kН2О =[Н+][ОН-][Н2О]–1
Поскольку количество диссоциированных молекул воды мало, концентрацию недиссоциированной воды считают постоянной, а её произведение [Н2О]-1×kН2О=[Н+][ОН-] даёт величину КН2О=[Н+][ОН-], зависящую от температуры и при 25°С равную 1,02×10-14моль2л-2, т.е. произведение [Н+][ОН-] =10-14. В нейтральных растворах [Н+][ОН-]=10-7. Логарифмируя приходим к уравнению –lg[Н+]= -lg[ОН-]=10-7. Приняв, что pН+=pOН-=7, получаем значение pН+=-lg[Н+], представляющее отрицательный десятичный логарифм молярной концентрации водородных ионов.
pН+ среды во многом определяет степень диссоциации растворённых в ней веществ и каталитическую активность ферментных систем[16], составляя одну из кондиций нормы реакции.
Таким образом, статистически оперируя стандартным набором элементарных участников и простейших реакций кооперации и превращений, эволюция невольно шла по пути термодинамической рентабельности усложнения структуры, устройства и организации материи. По мере усложнения и в связи с этим, автоматически была расширена вариантность (разнообразие) материального окружения в вероятностном режиме исполнения термодинамически выгодных процессов, положившая начало формирования прообраза некой внешней среды, появления каталитических принципов усовершенствования способов достижения целесообразности.
Усложнение материального мира, связанное с расширяющимся спектром взаимодействующих участников, «стремление» сложного к дальнейшей кооперации, неизбежно явило собой «необходимую целесообразность» усовершенствования исполнения (оптимизации) условий первичных «норм реакций» в формируемом неравновесии зарождающейся внешней среды. Пусть не сложных, но постоянно действующих систем регулирования и управления, действующих на обязательной основе принципа «обратной связи»: соотношений «плотностей» (концентраций) реагентов, продуктов взаимодействия и катализаторов в практически любых физико-химических процессах, происходящих в любой фазе их физического состояния: газообразной, жидкой, твёрдой.
Физическое регулирование
Поскольку эволюция «простого» фактически началась с усложнений «элементарного», т.е. с взаимодействий участников релятивистского мира, опосредованных одной только статистической вероятностью события и термодинамической рентабельностью усложнения, резонно предположить или даже быть уверенным в том, что прародители материального, живого и даже духовного, сохранили своё влияние, а возможно, и некую доминирующую роль в процессах и продуктах всей эволюции, на всех уровнях организации материи.
Факт этой «влиятельности» сам по себе не может быть оспорен, ибо подтверждён колоссальным количеством эмпирических исследований и специальных наблюдений [17] со времён профессора А.Л. Чижевского (1897-1964). Он может быть либо понят, либо не понят на уровне способностей современного знания, но отвергнутым не был и не станет никогда. Надо только не оставлять попыток обобщить и без того богатый фактический материал. Ведь одна только способность факторов прямого физического действия изменять скорость течения химических, а особенно каталитических процессов (фотохимические реакции) уже демонстрирует уникальное совершенство родоначального инструмента регулирования и управления сложнейшими физико-химическими и биофизическими процессами на непосредственном стыке живой и неживой природы. И такая способность связана:
1. с каталитическим эффектом уменьшения (или увеличения) энергии активации химических реакций под действием физических факторов: электрических, магнитных, электромагнитных, гравитационных и иных статических сил, позволяющих обеспечить доступность и/или когерентность (сродство) квантовых параметров участников физико-химического процесса (реагента, реагентов или катализатора) и/или параметрам сопровождающего и/или управляющего действия;
2. с сообщением реагентам и участникам химических превращений дополнительной энергии, способной преодолеть энергетический барьер через акцепт и возбуждение их молекулярных орбиталей под действием физических факторов релятивистского свойства;
3. с активацией неактивных и/или дезактивацией активных биологических катализаторов (ферментов), восстанавливающих и/или утрачивающих своё сродство к реагентам под действием физических факторов (эффекторов) релятивистского и нерелятивистского свойства.
Наиболее яркие тому примеры:
1. фотосинтез зелёных растений, демонстрирующий способность акцептовать элементарные релятивистские потоки фотонов адаптировав их частоты к частотам, соизмеримым с реальными скоростями химических преобразований;
2. эффекты фоторецепции, демонстрирующие глубоко избирательную и аналитическую способность акцепта информации, заключённой в релятивистском потоке фотонов различного спектра высоких частот через их трансформацию в возбуждение (потенциалы действия) на клеточном уровне нейронов;
3. эффекты трансформации ионного и протонного фосфорилирования, обеспеченные обратным сопряжением нерелятивистских диффузных процессов с «рождением» электрических и электромагнитных потоков, как представителей релятивистского мира и составляющих суть практически всех энергообразующих (энергоснабжающих) биохимических реакций.
Катализ
Катализом принято называть изменение скорости или возбуждение (провокацию)[18] химической реакции веществами (катализаторами), которые участвуют в реакции, но не входят в состав её конечных продуктов. Катализатор может многократно участвовать в промежуточном химическом взаимодействии с реагентами, а потому его количество обычно много меньше, чем собственно реагентов. По характеру действия катализатора различают:
1. положительный катализ, связанный с ускорением реакции;
2. отрицательный катализ, обеспечивающий замедление или ингибирование процесса.
По фазе размещения катализатора, каталитических систем и реагентов:
1. гомогенный катализ, когда реагент (реагенты) и катализатор находятся в одной фазе;
2. гетерогенный катализ, вызываемый катализаторами, находящимися или образующими самостоятельную фазу, отделённую от реагентов границей раздела;
3. мицеллярный катализ занимает промежуточное положение и связан с взаимодействием коллоидных частиц с реагентами в жидкой фазе;
4. гетерогенно-гомогенный катализ, при котором реакция начинается на поверхности катализатора, а продолжается в объёме.
По химической природе катализатора, неразрывно связанной с хронологией эволюционного процесса, отдельно выделяют:
1. катализ на металлах, оксидах и других простых веществах;
2. кислотно-основной катализ, при котором реакции протекают в присутствии диссоциирующих (ионизированных) кислот или оснований;
3. ферментативный катализ, составляющий отдельную группу ферментативных реакций, вызываемых биологическими катализаторами – ферментами и являющийся высшим эволюционным достижением в усовершенствовании химических процессов.
Мы считаем возможным расширить предлагаемую классификацию дополнительными, учитывающими способность факторов прямого физического действия изменять скорость течения химических, а особенно каталитических процессов, выделив отдельно:
1. «статический катализ», который связан с каталитическим эффектом уменьшения (или увеличения) энергии активации химических реакций под действием физических факторов: электрических, магнитных, электромагнитных, гравитационных, и иных статических сил, позволяющих обеспечить доступность и/или когерентность (сродство) квантовых параметров участников физико-химического процесса (реагента, реагентов или катализатора) и/или параметрам сопровождающего и/или управляющего действия;
2. «прямой квантовый катализ», связанный с сообщением реагентам дополнительной энергии, способной преодолеть энергетический барьер через акцепт и возбуждение их молекулярных орбиталей под действием физических факторов релятивистского свойства;
3. «опосредованный квантовый катализ», связанный с активацией неактивных и/или дезактивацией активных биологических катализаторов (ферментов), восстанавливающих и/или утрачивающих своё сродство к реагентам под действием физических факторов (эффекторов) релятивистского свойства.
Предлагаемое в пунктах 1 и 2 следовало бы разместить, согласно логики эволюции и собственного изложения, на первое место в приведенной классификации, поскольку «усовершенствование хаоса» рациональным усложнением материального мира имело именно это начало[19].
В связи с этим, следует напомнить и особенно подчеркнуть, что любое «усовершенствование» физико-химических процессов возможно осуществить только путём изменения скоростей реакций и эффективности взаимодействия реагентов посредством:
1. увеличения вероятности события,
2. уменьшения энергии активации процесса,
связанных с:
1. увеличением избирательности (сродства) к реагенту;
2. увеличением чувствительности катализатора;
4. увеличением избирательной чувствительности;
5. увеличением чувствительности к физическому фактору действия или сопровождения, обеспечивающий доступность или когерентность квантовых параметров участников физико-химического процесса (реагента, реагентов или катализатора) квантовым характеристикам действующих или сопровождающих факторов.
Однако, в любом случае, катализатор никогда не сдвигает химического равновесия в реагирующих системах. Он лишь в равной мере ускоряет или замедляет прямую и обратную реакции.
Таким образом, появление катализа позволило в большой мере реализовать второй путь такого «усовершенствования». Эволюция «научилась» «уменьшать энергию активации процессов». Одновременно, в связи с увеличением скоростей реакций в местах их исполнения, всё больше и больше концентрировалось вторичное неравновесие, не успевающее рассеяться во всеобщем хаосе энтропии. Так начали формироваться реальные предпосылки для более энергичных локальных коопераций, увеличивающих вероятность происходящих во времени и пространстве событий, т.е. приступить к реализации первого из названных выше путей в «усовершенствовании» физико-химических процессов.
Дальнейшая перспектива увеличения вероятности событий могла быть связана только с оптимизацией условий эффективного исполнения физико-химических преобразований. Эволюция развития «требовала» энергетически выгодной и статистически предопределенной тактики усложнений:
1. изменение дистантности (расстояния) для участников (реагентов),
2. их компартментализации,
3. кооперации,
4. уплотнения,
5. физическое сближения самих реагентов,
6. реактивных центров катализаторов,
7. формирования каталитических систем.
Т.е. всех тех действий, которые увеличивали бы статистическую вероятность события (взаимодействия), сопряженного с минимизацией энергетических затрат, повышением их эффективности.
Таким образом, статистическая вероятность последовательно, шаг за шагом была выведена историей эволюции на качественно новый уровень управления при использовании практически одного и того же набора инструментов в борьбе за оптимизацию их эффективности и рентабельности.
Вот почему сама последовательность исторических событий рационального усложнения элементарного и простого требовала «концентрации сил» в замкнутых объёмах гомеостатических компартментов. Но для этого сначала нужна была жидкая среда, усиливающая эффект неравновесия. Затем необходимо было дождаться логического появления в ней органических соединений, мембран, мембранных белково-липидных коопераций, ставших структурно-функциональной базой для рождения и усовершенствования ферментативного катализа.
Биологический (ферментативный) катализ
Таким образом, поднимаясь вверх по лестнице эволюционного онтогенеза, нам удалось отследить закономерную неизбежность рождения наиболее эффективного инструмента регулирования (управления) и ускорения (оперативного исполнения управляющих требований) процессов химических превращений в общем балансе обмена веществом, энергией и информацией неравновесных систем – ферментативный (или биологический) катализ. Но прежде, чем приступить к описанию особенностей ферментативного катализа и путей его эволюционного усовершенствования, очень важно вспомнить, что эволюция невольно должна была идти по пути усложнения не только структуры и организации самой материи, но и её материального окружения. Это связано с тем, что создание любого сложного, эффективного, а значит чувствительного инструмента под давлением причинно следственных обстоятельств требует оптимизации условий его «хранения и защиты». Оно нуждается в гомеостазе. А уже тем более, если речь идёт о таком инструменте, каким является ферментативный катализ.
Сегодня известно более 2000 ферментов, которые согласно рекомендациям специальной Комиссии по ферментам Международного биохимического союза подразделяются на 6 классов в зависимости от характера катализируемых ими реакций:
1. оксидоредуктазы – класс ферментов, катализирующих окислительно-восстановительные реакции. Окисляемыми субстратами могут быть соединения, содержащие спиртовую, альдегидную, кетонную или другие группы, а также восстанавливаемые формы никотинамидных коферментов (НАД и НАДФ);
2. трансферазы – класс ферментов, катализирующих перенос групп атомов от одной молекулы к другой. Различают трансферазы, катализирующие перенос одноуглеродных, ацильных и гликозильных остатков, фосфатных групп, остатков, содержащих азот и др.;
3. гидролазы – класс ферментов, катализирующих гидролиз связей между атомом углерода и гетероатомом, в частности – пептидных, амидных, гликозидных, сложноэфирных связей;
4. лиазы – класс ферментов, катализирующих негидролитическое отщепление (С–О; C–S; C–N; C-галоген) атомов или групп атомов от субстрата с образованием двойных связей и обратные реакции присоединения по двойным связям;
5. изомеразы – класс ферментов, катализирующих изомеризацию, иногда – перенос какой-либо группы от одного участка молекулы к другому (мутазы);
6. лигазы (синтетазы) – класс ферментов, катализирующих присоединение друг к другу двух молекул (С–О; C–S; C–N; C-C), сопряжённое с расщеплением пирофосфатной связи.
Благодаря своим доведённым до совершенства свойствам, ферментативный катализ характеризуется чрезвычайно высокой активностью и специфичностью (селективностью). Это связано, прежде всего, с исполнением названных выше требований изменения дистантности (расстояния) участников, их компартментализации, кооперации, уплотнения, физического сближения реагентов и реактивных центров.
1. Одно только образование так называемого «активного комплекса Михаэлиса»[20] позволяет увеличить скорость катализируемой реакции в 107 и более раз;
2. Появившаяся в результате сближения возможность полифункционального характера химического взаимодействия[21] позволяет добиться ускорения реакции еще в 103 и более раз;
3. Эффекты микросреды в области активного центра могут дополнительно обеспечить ускорение реакции еще в 105 раз.
Таким образом, исполнение условий изменения дистантности, стехиометрического соответствия активного центра фермента и субстрата, обеспечивают суммарное ускорение катализируемых реакций приблизительно в 108-1012-1015 раз[22]. Вот почему сама динамика исторических событий рационального усложнения элементарного и простого в рациональное сложное требовала «концентрации сил» в замкнутых объёмах гомеостатических компартментов. Но ведь и этим не смогла ограничиться эволюция усовершенствования эффективности и действенности своих инструментов управления.
Сформированные условия «первичного» в биологическом катализе (гомеорстаз, комплекс Михаэлиса, стехиометрическое сродство) сами определили (индуцировали) путь для такого усовершенствования:
1. через сложную, но идеальную для регулирования (изменения активностей) композицию вторичной (спираль), третичной (двойная спираль), четвертичной (глобула) и пятеричной (белково-липидный композит мембран) структуры организации материи;
2. через электростатическое сродство сложных пространственных конфигураций каталитических центров (стехиометрии) молекулярных структур биологических катализаторов (ферментов) к пространственной конфигурации молекул реагентов (матричного типа);
3. через электростатическое сродство молекул конечных продуктов после завершения каждого предыдущего этапа катализа к конфигурации каталитического центра следующего фермента в цепи последовательных химических превращений.
Сам ход последовательных катализируемых реакций предопределил путь усовершенствования биологического катализа формированием последовательных ферментных цепей в единый структурно-функциональный комплекс, соответствующий последовательностям необходимых химических преобразований, требующих такого катализа. При этом необходимость рационального и последовательного катализа была предопределена не только его очевидной целесообразностью и рациональностью, она была обустроена силами межмолекулярных взаимодействий электростатического свойства, заставивших шаг за шагом избирательно «выстроить» необходимые ферменты в каталитический конвейер требуемого биохимического процесса. Относительная же слабость сил межмолекулярного взаимодействия (в основном, Ван-дер-ваальсовых, ионных, электростатических) гарантировала высокую мобильность в изменениях пространственных конфигураций (конформаций) макромолекул без потери ими основной структуры (первичной, вторичной и третичной), обеспеченной более прочными связями межатомных взаимодействий. Так был рождён совершеннейший инструмент стехиометрического сродства (и/или потери такого сродства) взаимодействующих реагентов.
Вместе с тем, усложнение структуры отдельных участников вплоть до образования серьёзных композитов, сочетанное со стабилизацией среды их функционирования, не могло не отразиться на параметрах их чувствительности к прямым физическим воздействиям (температура, давление, электромагнитные излучения, влияние статических и других физических полей и потоков).
Вместо заключения
На основании вышеизложенного, наверное, уже не так трудно назвать искомые элементы общего на уровне участников субстратных, ионных, физических и иных взаимоотношений, как того требовал избранный нами подход к изложению «теории адаптации и адаптивной регуляции гомеостатических систем» и единого гомеостатического устройства.
Поэтому вооружившись ответами на сформулированные в начале изложения вопросы: кто есть участники, партнёры, исполнители гомеостатических отношений, каково их инструментальное обеспечение в общем балансе обмена веществом, энергией и информацией, каков мотив этих отношений и как он реализуется, можно позволить себе дальше двигаться вверх по иерархической лестнице.
Вопрос: кто является участниками названных отношений?
Ответ: вещества самого широко спектра: от элементарных частиц, атомов, молекул и ионов до сложнейших органических соединений и их композитов, включая каталитические системы.
Вопрос: кого можно назвать «партнером» в этих отношениях?
Ответ: любой субъект или допустимый тип действия, способный вызвать изменение состояния (возбуждение) участников из числа названных (от элементарных частиц, атомов, молекул и ионов до сложнейших органических соединений и их композитов).
Вопрос: каков ожидаемый эффект такого действия?
Ответ: изменение состояния, реактивности или качества названного спектра участников как следствие возбуждения (от элементарных частиц, атомов, молекул и ионов до сложнейших органических соединений и их композитов).
Таким образом:
1. Участниками реальных отношений являются все представители материального мира от элементарных частиц, атомов, молекул и ионов до сложнейших органических и неорганических соединений и их композитов;
2. объектом регулирования являются практически все отношения участников материального мира от элементарных частиц, атомов, молекул и ионов до сложнейших органических и неорганических соединений и их композитов;
3. мотивом регулирования названных отношений участников является термодинамическая рентабельность;
4. способом реализации отношений участников являются практически все химические, физико-химические и физические процессы и преобразования, сопровождающиеся термодинамической рентабельностью;
5. базовым средством осуществления таких отношений является общий баланс (регламент) этих отношений, составляющих суть нормы реакции;
6. составляющими нормы реакции на уровне отношений участников являются: скорость реакции, каталитическое обеспечение, условия её исполнения (среду, температуру, давление);
7. инструментами управления скорости реакций (отношений участников) являются: субстратное, ионное, каталитическое и физическое регулирование.
Таким образом, совершенно очевидно, что сам ход эволюции «требовал» сохранить и хоть как-то защитить зарождающееся неравновесие в попытке утвердить выгодное ей «производство энтропии». В связи с этим, сама логика усложнения материального подготовила всё для появления замкнутых пространств, физических барьеров, естественных компартментов, ограждающих эти пространства от внешних случайностей. И такими компартментами стали органоиды, органеллы, растительные и животные клетки сначала примитивной, а затем и высокоорганизованной (живой) материи.
Глава 3
Гомеостаз органоидов и органелл
[1] Энергия, необходимая для осуществления эффективного столкновения молекул реагентов, приводящего к химической реакции. Энергия активации определяется как минимальная величина энергии, которой должна обладать молекула для вступления в реакцию
[2] Для реакций в газах – константа летучести (К f)
[3] Константа равновесия может быть выражена через парциальные давления pi, молярные концентрации ci, диссоциацию di реагентов и продуктов и обозначаться соответственно Кр (Кр º Кf) или Кc (Кc º Кd)
[4] DG° можно найти из:
1. констант равновесия (DG° = -RT ln Кр);
2. окислительно-восстановительных потенциалов (DG° = -nFEвосст/ок);
3. табличных значений энтальпии и энтропии (DG° = DH°- TDS°);
4. разности DG° реагирующих веществ и DG°продуктов реакции (DG° = G°продукты- G°реагенты)
[5] исключение составляет прямая фотоионизация, связанная с распадом молекулы вещества на его ион и электрон. От других фотохимических реакций фотоионизация отличается тем, что процесс происходит непосредственно при поглощении фотона без возбуждения молекулярных орбиталей (электронных состояний). Обычно наблюдается в газовой среде, когда энергия фотонов превышает энергию ионизации.
[6] Двухквантовые реакции – возникают в результате поглощения двух фотонов света, причём второй квант поглощается в одном из низших электронно-возбуждённых состояний молекулы
[7] Багдарасьян Х.С. Двухквантовая фотохимия, М.1976
[8] обратимые процессы фотоизомеризации лежат в основе фотохромизма (изменение окраски)
[9] Nernst W. Die theoretischen und experimentallen Grundlagen des neuen Warmesatzes, Halle (Saale), W., 1918; Nernst W. Experimental and theoretical applications of thermodynamics to chemistry, New Haven, 1913; Nernst W. Theoretische Chemie vom Standpunkte der Avogadroschen Regel und Thermodynamik, 15 Aufl., Stuttgart, 1926; В.Нернст Теоретическая химия с точки зрения закона Авогадро и термодинамики, СПБ, 1904; В.Нернст Основания высшей математики, М,: 1907 (совместно с Шенфлиссом)
[10] мономолекулярная реакция – одинаковые молекулы претерпевает расщепление или перегруппировку или кооперируют
[11] Эта константа численно равна скорости реакции при молярных концентрациях, равных единице.
[12] Муссил Я., Новакова О., Кунц К Современная биохимия в схемах, М, Мир, 1984, 215 с.
[13] позже, при рассмотрении механизмов регулирования на уровне планетарного гомеостаза, мы будем иметь возможность вернуться к названным принципам более подробно, рассматривая возможности управления на уровне Форбуш взаимодействия планетарной биосферы с высшим для неё гомеостатическим иерархом, Космосом.
[14] Формульно представлена условная иллюстрация, регулируемая константой равновесия: реагенты - продукт
[15] По М.Дьюара и Р.Догерти (1977)
[16] Мецлер Д. Биохимия, М, Мир, 1980; Ленинджер А. Основы биохимии, М, Мир, 1985
[17] Агулова Л.П. Принципы адаптации биологических систем к космогеофизическим факторам/Биофизика, 1998, том 43, вып. 4, с.571-574); Доронин В.Н., Парфентьев В.А., Тлеулин С.Ж., Намвар Р.А., Сомсиков В.М., Дробжев В.И., Чемерис А.В. Влияние вариаций геомагнитного поля и солнечной активности на физиологические показатели человека./В жрун. Биофизика, 1998, том 43, вып. 4, с.647-653; Корнелиссен Ж., Халберг Ф., Бреус Т.К., Ватанабе И., Сотерн Р.Б., Хаус Е., Клейтмен Е., Вендт Х.В., Бинхам К. О проблеме происхождения биологической недели по данным о вариациях ритма частоты сердечных сокращений у людей в цикле солнечной активности/В журн. Биофизика, 1998, том 43, вып. 4, с.666-669; Мак-Фадден Б.Дж., Джонсон Д.С., Манн С., Нессон М.Х., Ловенстам Х.А., Прести Д.Е., Пэрри Э., Бауэр Г., Дайзон Э. И многие другие в книге Биогенный магнетит и магниторецепция. М.: Мир, 1989; Швецов Ю.П., Новиков В.В., Фесенко Е.Е., Чернов А.П., Иванов В.А. Молекулярные механизмы биологического действия слабых магнитных полей/В журн. Биофизика, 1998, ноябрь-декабрь, том 43, номер 6, с.977-980; Карнаухов А.В. Диссипативный резонанс и его роль в механизмах действия э/м излучения на биологические и физико-химические системы/В журн. Биофизика, 1997, том 42, вып 4, с.971-978 и т.д.
[18] В химии принято пользоваться термином «возбуждение» химической реакции. Мы умышленно акцентируем внимание на этом термине, поскольку пользовали и пользуем его для характеристики состояния участников гомеостатического мира: «возбуждение молекулярных орбиталей», «возбуждение атомов», «возбуждение молекул», «возбуждение акцепторных зон»», «возбуждение гомеостатических систем». Таким образом, мы считаем, что термин «возбуждение» применимый к химическим реакциям, можно квалифицировать как частный случай «возбуждения инструментов управления» в любой гомеостатической системе.
[19] именно его следовало бы назвать первым прецедентом рождения энтропии по И.Р. Пригожину
[20] композит фермента и субстрата
[21] атака активного центра фермента сразу несколькими каталитическими группами
[22] Кретович В.Л. Введение в энзимологию, 2-е издание, М., 1974; Березин И.В., Мартинек К. Основы физической химии ферментативного катализа, М., 1977; Клесов А.А., Березин И.В. Ферментативный катализ, ч.1, М., 1980; Клесов А.А Ферментативный катализ, БЭС, Химия, М., 1998, с.617; Фершт Э. Структура и механизмы действия ферментов, м., 1980; Диксон М, Уэбб Э. Ферменты, т.1-3, М., 1982; Антонов В.К. Ферменты, БЭС, Химия, М., 1998, с.617-618
Оглавление Введение Часть 1 Часть 2 Часть 3 Заключение
Глава 1 Глава 2 Глава 3 Глава 4 Глава 5 Глава 6 Глава 7 Литература